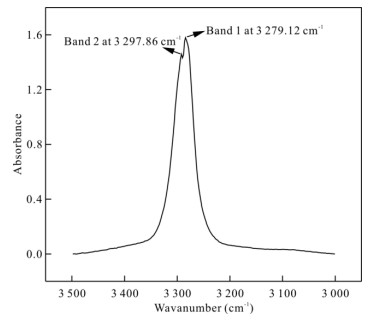
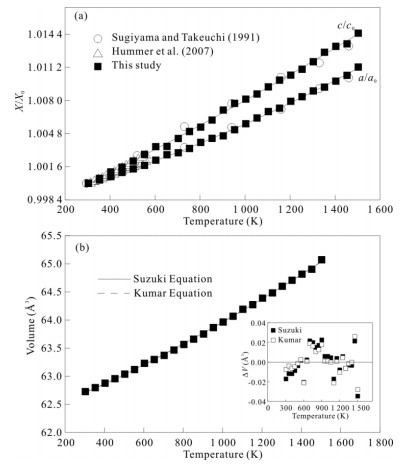
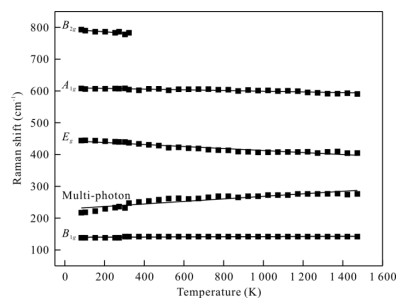
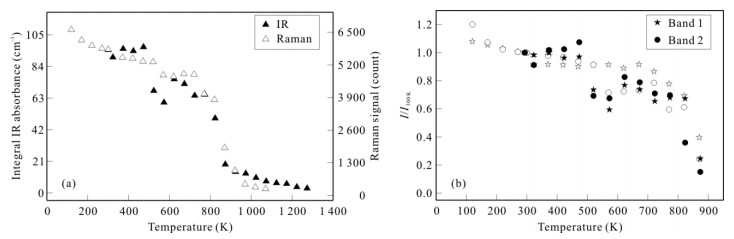
| Citation: | Sha Wang, Jinhua Zhang, Joseph R Smyth, Junfeng Zhang, Dan Liu, Xi Zhu, Xiang Wang, Yu Ye. Crystal Structure, Thermal Expansivity and High-Temperature Vibrational Spectra on Natural Hydrous Rutile. Journal of Earth Science, 2020, 31(6): 1190-1199. doi: 10.1007/s12583-020-1351-5

|
| Arlt, T., Bermejo, M., Blanco, M. A., et al., 2000. High-Pressure Polymorphs of Anatase TiO2. Physical Review B, 61(21):14414-14419. https://doi.org/10.1103/physrevb.61.14414 |
| Balachandran, U., Eror, N. G., 1982. Raman Spectra of Titanium Dioxide. Journal of Solid State Chemistry, 42(3):276-282. https://doi.org/10.1016/0022-4596(82)90006-8 |
| Bromiley, G. D., Hilairet, N., 2005. Hydrogen and Minor Element Incorporation in Synthetic Rutile. Mineralogical Magazine, 69(3):345-358. https://doi.org/10.1180/0026461056930256 |
| Bromiley, G. D., Shiryaev, A. A., 2006. Neutron Irradiation and Post-Irradiation Annealing of Rutile (TiO2-x):Effect on Hydrogen Incorporation and Optical Absorption. Physics and Chemistry of Minerals, 33(6):426-434. https://doi.org/10.1007/s00269-006-0087-9 |
| Bromiley, G. D., Hilairet, N., Mccammon, C., 2004. Solubility of Hydrogen and Ferric Iron in Rutile and TiO2 (Ⅱ):Implications for Phase Assemblages during Ultrahigh-Pressure Metamorphism and for the Stability of Silica Polymorphs in the Lower Mantle. Geophysical Research Letters, 31(4):L04610. https://doi.org/10.1029/2004gl019430 |
| Cao, Y. T., Liu, L., Yang, W. Q., et al., 2019. Reconstruction the Process of Partial Melting of the Retrograde Eclogite from the North Qaidam, Western China:Constraints from Titanite U-Pb Dating and Mineral Chemistry. Journal of Earth Science, 30(6):1166-1177. https://doi.org/10.1007/s12583-019-1253-6 |
| Cromer, D. T., Mann, J. B., 1968. X-Ray Scattering Factors Computed from Numerical Hartree-Fock Wave Functions. Acta Crystallographica Section A, 24(2):321-324. https://doi.org/10.1107/s0567739468000550 |
| Deer, W. A., Howie, R. A., Zussman, J., 1963. An Introduction to the Rock-Forming Minerals. Journal of Geology, 71:534-536. https://doi.org/10.1086/626928 |
| Dolomanov, O. V., Blake, A. J., Champness, N. R., et al., 2003. OLEX:New Software for Visualization and Analysis of Extended Crystal Structures. Journal of Applied Crystallography, 36(5):1283-1284. https://doi.org/10.1107/s0021889803015267 |
| Downs, R. T., Bartelmehs, K. L., Gibbs, G. V., et al., 1993. Interactive Software for Calculating and Displaying X-Ray or Neutron Powder Diffractometer Patterns of Crystalline Materials. American Mineralogist, 78:1104-1107. https://doi.org/10.1029/93jb01427 |
| Fei, Y., 1995. Thermal Expansion. In: Ahrens, J. T., ed., Mineral Physics and Crystallography. American Geophysical Union, Washington. 29-44 |
| Foley, S. F., Barth, M. G., Jenner, G. A., 2000. Rutile/Melt Partition Coefficients for Trace Elements and an Assessment of the Influence of Rutile on the Trace Element Characteristics of Subduction Zone Magmas. Geochimica et Cosmochimica Acta, 64(5):933-938. https://doi.org/10.1016/s0016-7037(99)00355-5 |
| Guo, H. H., 2017. In-situ Infrared Spectra of OH in Rutile up to 1 000 ℃. Physics and Chemistry of Minerals, 44(8):547-552. https://doi.org/10.1007/s00269-017-0881-6 |
| Hammer, V. M. F., Beran, A., 1991. Variations in the OH Concentration of Rutiles from Different Geological Environments. Mineralogy and Petrology, 45(1):1-9. https://doi.org/10.1007/bf01164498 |
| Hara, Y., Nicol, M., 1979. Raman Spectra and the Structure of Rutile at High Pressures. Physica Status Solidi B, 94(1):317-322. https://doi.org/10.1002/pssb.2220940137 |
| Hazen, R. M., Finger, L. W., 1981. Bulk Moduli and High-Pressure Crystal Structures of Rutile-Type Compounds. Journal of Physics and Chemistry of Solids, 42(3):143-151. https://doi.org/10.1016/0022-3697(81)90074-3 |
| Hemley, R. J., Mao, H. K., Chao, E. C. T., 1986. Raman Spectrum of Natural and Synthetic Stishovite. Physics and Chemistry of Minerals, 13(5):285-290. https://doi.org/10.1007/bf00308345 |
| Henderson, C. M. B., Knight, K. S., Lennie, A. R., 2009. Temperature Dependence of Rutile (TiO2) and Geikielite (MgTiO3) Structures Determined Using Neutron Powder Diffraction. The Open Mineralogy Journal, 3(1):1-11. https://doi.org/10.2174/1874456700903010001 |
| Holland, T. J. B., Redfern, S. A. T., 1997. Unit Cell Refinement from Powder Diffraction Data:The Use of Regression Diagnostics. Mineralogical Magazine, 61(404):65-77. https://doi.org/10.1180/minmag.1997.061.404.07 |
| Howard, C. J., Sabine, T. M., Dickson, F., 1991. Structural and Thermal Parameters for Rutile and Anatase. Acta Crystallographica Section B Structural Science, 47(4):462-468. https://doi.org/10.1107/s010876819100335x |
| Hummer, D. R., Heaney, P. J., Post, J. E., 2007. Thermal Expansion of Anatase and Rutile between 300 and 575 K Using Synchrotron Powder X-Ray Diffraction. Powder Diffraction, 22:352-357. https://doi.org/10.1154/1.2790965 |
| Isaak, D. G., Carnes, J. D., Anderson, O. L., et al., 1998. Elasticity of TiO2 Rutile to 1 800 K. Physics and Chemistry of Minerals, 26(1):31-43. https://doi.org/10.1007/s002690050158 |
| Johnson, O. W., Ohlsen, W. D., Kingsbury, P. I., 1968. Defects in Rutile Ⅲ. Optical and Electronic Properties of Impurities and Charge Carriers. Physical Review, 175:1102-1109. https://doi.org/10.1103/physrev.185.1230.2 |
| Johnson, O. W., DeFord, J., Shaner, J. W., 1973. Experimental Technique for the Precise Determination of H and D Concentration in Rutile (TiO2). Journal of Applied Physics, 44(7):3008-3012. https://doi.org/10.1063/1.1662697 |
| Klemme, S., Blundy, J. D., Wood, B. J., 2002. Experimental Constraints on Major and Trace Element Partitioning during Partial Melting of Eclogite. Geochimica et Cosmochimica Acta, 66(17):3109-3123. https://doi.org/10.1016/s0016-7037(02)00859-1 |
| Koudriachova, M. V., de Leeuw, S. W., Harrison, N. M., 2004. First-Principles Study of H Intercalation in Rutile TiO2. Physical Review B, 70(16):165421. https://doi.org/10.1103/physrevb.70.165421 |
| Kumar, M., 1995. High Pressure Equation of State for Solids. Physica B:Condensed Matter, 212(4):391-394. https://doi.org/10.1016/0921-4526(95)00361-c |
| Kumar, M., 1996. Application of High Pressure Equation of State for Different Classes of Solids. Physica B:Condensed Matter, 217(1/2):143-148. https://doi.org/10.1016/0921-4526(95)00448-3 |
| Kumar, M., 2003. Thermoelastic Properties of Minerals. Physics and Chemistry of Minerals, 30:556-558. https://doi.org/10.1007/s00269-003-0344-0 |
| Lan, T., Tang, X. L., Fultz, B., 2012. Phonon Anharmonicity of Rutile TiO2 Studied by Raman Spectrometry and Molecular Dynamics Simulations. Physical Review B, 85(9):094305. https://doi.org/10.1103/physrevb.85.094305 |
| Li, K. Y., Xue, D. F., 2006. Estimation of Electronegativity Values of Elements in Different Valence States. The Journal of Physical Chemistry A, 110(39):11332-11337. https://doi.org/10.1021/jp062886k |
| Libowitzky, E., 1999. Correlation of O-H Stretching Frequencies and O-H…O Hydrogen Bond Lengths in Minerals. Monatshefte für Chemie, 130(8):1047-1059. https://doi.org/10.1007/bf03354882 |
| Litasov, K. D., Kagi, H., Shatskiy, A., et al., 2007. High Hydrogen Solubility in Al-Rich Stishovite and Water Transport in the Lower Mantle. Earth and Planetary Science Letters, 262(3/4):620-634. https://doi.org/10.1016/j.epsl.2007.08.015 |
| Lucassen, F., Koch-Muller, M., Taran, M., et al., 2012. Coupled H and Nb, Cr, and V Trace Element Behavior in Synthetic Rutile at 600 ℃, 400 MPa and Possible Geological Application. American Mineralogist, 98(1):7-18. https://doi.org/10.2138/am.2013.4183 |
| Maldener, J., Rauch, F., Gavranic, M., et al., 2001. OH Absorption Coefficients of Rutile and Cassiterite Deduced from Nuclear Reaction Analysis and FTIR Spectroscopy. Mineralogy and Petrology, 71(1/2):21-29. https://doi.org/10.1007/s007100170043 |
| Mammone, J. F., Sharma, S. K., Nicol, M., 1980. Raman Study of Rutile (TiO2) at High Pressures. Solid State Communications, 34(10):799-802. https://doi.org/10.1016/0038-1098(80)91055-8 |
| Meagher, E. P., Lager, G. A., 1979. Polyhedral Thermal Expansion in the TiO2 Polymorphs:Refinement of the Crystal Structure of Rutile and Brookite at High Temperature. The Canadian Mineralogist, 17:77-85 http://www.researchgate.net/publication/299160965_Polyhedral_thermal_expansion_in_the_TiO2_polymorphs_Refinement_of_the_crystal_structures_of_rutile_and_brookite_at_high_temperature |
| Miao, Y. F., Pang, Y. W., Ye, Y., et al., 2019. Crystal Structures and High-Temperature Vibrational Spectra for Synthetic Boron and Aluminum Doped Hydrous Coesite. Crystals, 9(12):642. https://doi.org/10.3390/cryst9120642 |
| Ming, L. C., Manghnani, M. H., 1979. Isothermal Compression of TiO2 (Rutile) under Hydrostatic Pressure to 106 kbar. Journal of Geophysical Research, 84(B9):4777-4779. https://doi.org/10.1029/jb084ib09p04777 |
| Mookherjee, M., Redfern, S. A. T., Zhang, M., 2001. Thermal Response of Structure and Hydroxyl Ion of Phengite-2M1:An in situ Neutron Diffraction and FTIR Study. European Journal of Mineralogy, 13(3):545-555. https://doi.org/10.1127/0935-1221/2001/0013-0545 |
| Nie, J. Z., Liu, Y. C., Yang, Y., 2018. Phase Equilibria Modeling and P-T Evolution of the Mafic Lower-Crustal Xenoliths from the Southeastern Margin of the North China Craton. Journal of Earth Science, 29(5):1236-1253. https://doi.org/10.1007/s12583-018-0849-6 |
| Pawley, A. R., McMillan, P. F., Holloway, J. R., 1993. Hydrogen in Stishovite, with Implications for Mantle Water Content. Science, 261(5124):1024-1026. https://doi.org/10.1126/science.261.5124.1024 |
| Porto, S. P. S., Fleury, P. A., Damen, T. C., 1967. Raman Spectra of TiO2, MgF2, ZnF2, FeF2 and MnF2. Physical Review, 154(2):522-526. https://doi.org/10.1103/physrev.154.522 |
| Rao, K. V. K., Naidu, S. V. N., Iyengar, L., 1970. Thermal Expansion of Rutile and Anatase. Journal of the American Ceramic Society, 53(3):124-126. https://doi.org/10.1111/j.1151-2916.1970.tb12051.x |
| Rossman, G. R., Smyth, J. R., 1990. Hydroxyl Content of Accessory Minerals in Mantle Eclogites and Related Rocks. American Mineralogist, 75:775-780 http://ci.nii.ac.jp/naid/80005531407 |
| Samara, G. A., Peercy, P. S., 1973. Pressure and Temperature Dependence of the Static Dielectric Constants and Raman Spectra of TiO2 (Rutile). Physical Review B, 7(3):1131-1148. https://doi.org/10.1103/physrevb.7.1131 |
| Sato, Y., 1977. Equation of State of Mantle Minerals Determined through High-Pressure X-Day Study. High Pressure Research Applications in Geophysics, (1977):307-323. https://doi.org/10.1016/b978-0-12-468750-9.50028-0 |
| Saxena, S. K., Chatterjee, N., Fei, Y., et al., 1993. Thermodynamic Data on Oxides and Silicates:An Assessed Data Set Based on Thermochemistry and High Pressure Phase Equilibrium. Springer-Verlag, Berlin, Heidelberg, New York |
| Sheng, Y. M., Xia, Q. K., Hao, Y. T., 2007. Water in Rutiles from UHP Eclogites in the Dabie Orogen. Acta Petrologica et Mineralogica, 26:269-274 (in Chinese with English Abstract) http://en.cnki.com.cn/Article_en/CJFDTOTAL-YSKW200703009.htm |
| Soffer, B. H., 1961. Studies of the Optical and Infrared Absorption Spectra of Rutile Single Crystals. The Journal of Chemical Physics, 35(3):940-945. https://doi.org/10.1063/1.1701242 |
| Song, Y. R., Jin, Z. M., 2002. Nanometer-Sized UHP Rutile:Tracing the Depth of Continental Deep Subduction. Earth Science Frontiers, 9:267-272 (in Chinese with English Abstract) http://en.cnki.com.cn/Article_en/CJFDTOTAL-DXQY200204008.htm |
| Su, W., Li, J. L., Mao, Q., et al., 2018. Rutile in HP Rocks from the Western Tianshan, China:Mineralogy and Its Economic Implications. Journal of Earth Science, 29(5):1049-1059. https://doi.org/10.1007/s12583-018-0848-7 |
| Sugiyama, K., Takéuchi, Y., 1991. The Crystal Structure of Rutile as a Function of Temperature up to 1 600℃. Zeitschrift für Kristallographie-Crystalline Materials, 194(1/2/3/4):305-313. https://doi.org/10.1524/zkri.1991.194.14.305 |
| Suzuki, I., 1975. Thermal Expansion of Periclase and Olivine, and Their Anharmonic Properties. Journal of Physics of the Earth, 23(2):145-159. https://doi.org/10.4294/jpe1952.23.145 |
| Suzuki, I., Okajima, S. I., Seya, K., 1979. Thermal Expansion of Single-Crystal Manganosite. Journal of Physics of the Earth, 27(1):63-69. https://doi.org/10.4294/jpe1952.27.63 |
| Swope, R. J., Smyth, J. R., Larson, A. C., 1995. H in Rutile-Type Compounds:I. Single-Crystal Neutron and X-Ray Diffraction Study of H in Rutile. American Mineralogist, 80(5/6):448-453. https://doi.org/10.2138/am-1995-5-604 |
| Tokonami, M., 1965. Atomic Scattering Factor for O2-. Acta Crystallographica, 19(3):486-486. https://doi.org/10.1107/s0365110x65003729 |
| Touloukian, Y. S., Kirby, R. K., 1977. Thermophysical Properties of Matter; Volume 13:Thermal Expansion; Nonmetallic Solids. IFI/Plenum, New York, Washington |
| Vlassopoulos, D., Rossman, G. R., Haggerty, S. E., 1993. Coupled Substitution of High and Minor Elements in Rutile and the Implications of High OH Contents in Nb-and Cr-Rich Rutile from the Upper Mantle. American Mineralogist, 78:1181-1191 http://ammin.geoscienceworld.org/content/78/11-12/1181 |
| Wang, X., Xu, X. X., Ye, Y., et al., 2019. In-situ High-Temperature XRD and FTIR for Calcite, Dolomite and Magnesite:Anharmonic Contribution to the Thermodynamic Properties. Journal of Earth Science, 30(5):964-976. https://doi.org/10.1007/s12583-019-1236-7 |
| Xie, Z. J., Liu, X. W., Jin, Z. M., et al., 2020. Microstructures and Phase Transition in Omphacite:Constraints on the P-T Path of Shuanghe Eclogite (Dabie Orogen). Journal of Earth Science, 31(2):254-261. https://doi.org/10.1007/s12583-019-1279-9 |
| Xiong, X. L., Adam, J., Green, T. H., 2005. Rutile Stability and Rutile/Melt HFSE Partitioning during Partial Melting of Hydrous Basalt:Implications for TTG Genesis. Chemical Geology, 218(3/4):339-359. https://doi.org/10.1016/j.chemgeo.2005.01.014 |
| Xiong, X. L., Keppler, H., Audétat, A., et al., 2011. Partitioning of Nb and Ta between Rutile and Felsic Melt and the Fractionation of Nb/Ta during Partial Melting of Hydrous Metabasalt. Geochimica et Cosmochimica Acta, 75(7):1673-1692. https://doi.org/10.1016/j.gca.2010.06.039 |
| Yang, Y., Xia, Q., Feng, M., et al., 2011. In situ FTIR Investigations at Varying Temperatures on Hydrous Components in Rutile. American Mineralogist, 96(11/12):1851-1855. https://doi.org/10.2138/am.2011.3826 |
| Zack, T., Kronz, A., Foley, S. F., et al., 2002. Trace Element Abundances in Rutiles from Eclogites and Associated Garnet Mica Schists. Chemical Geology, 184(1/2):97-122. https://doi.org/10.1016/s0009-2541(01)00357-6 |
| Zaffiro, G., Angel, R. J., Alvaro, M., 2019. Constraints on the Equations of State of Stiff Anisotropic Minerals:Rutile, and the Implications for Rutile Elastic Barometry. Mineralogical Magazine, 83(3):339-347. https://doi.org/10.1180/mgm.2019.24 |
Figures(10) / Tables(1)
Copyright © 2013-2020 Journal of Earth Science 鄂ICP备15021562号-2
Tel: +86-27-67885075 Fax: +86-27-67885075 E-mail: xbb@cug.edu.cn
Address: Editorial Office of Journal, China University of Geosciences, Yujiashan, Wuhan, Hubei 430074, P. R. China
Supported by:
Beijing Renhe Information Technology Co. Ltd
E-mail:
info@rhhz.net



 DownLoad:
DownLoad: